
Introduction
Lymphocystis disease is a common, chronic and benign infection that results in hypertrophy of fibroblastic cells in the dermis connective tissue (Samalecos 1986). The disease was characterised by nodules usually occurring on the skin and fins, as well as sometimes in internal organs and gills (Anders 1989). The disease has a worldwide distribution and has been reported from more than 125 marine and freshwater fish species (Borrego et al. 2015).
The causative agent of the disease is the lymphocystis disease virus (LCDV) (Wolf 1988). While detectability of the virus in untreated seawater takes 7-15 days depending on the water temperature, this period can be up to 240 days dependent on the water temperature, water quality and the treatment performed (Leiva-Rebollo et al. 2021). Viral transmission is horizontally caused by direct contact and via water. In addition, physical traumas on the skin facilitate transmission (Wolf 1988). It is also thought that the virus can be transmitted by feeding (Cano et al. 2012; Valverde et al. 2019). Vertical transmission is thought to be via viruses on the surface of untreated eggs (Cano et al. 2012).
LCDV has icosahedral symmetrical morphology with virions measuring 150-350 nm diameter containing a double-stranded linear DNA genome (Berthiaume, Alain, and Robin 1984; Jancovich et al. 2012). LDCV is a member of the Lymphocystivirus genus of the Iridoviridae family (Jancovich et al. 2012). The Lymphocystivirus genus contains four species named LCDV 1, 2 (c), 3 (SA) and 4 (WC): they were isolated from European flounder, Japanese flounder, gilthead seabream and whitemouth croaker, respectively (ICVT, 2020). Moreover, these species have different genome sizes of 102,653, 186,250, 208,501, and 211,086 bp, respectively (Tidona and Darai 1997; Zhang et al. 2004; López-Bueno et al. 2016; Doszpoly et al. 2020).
The major capsid protein (MCP) gene, which is a relatively conserved region in iridoviruses, was reported to be a suitable target for phylogenetic studies (Tidona et al. 1998). In the phylogenetic analysis of LCDV with the MCP gene as a target, LCDV isolates reported from 11 different species were divided into nine genotypes (Palmer, Hogan, and van den Heuvel 2012). According to this classification: LCDV-1 European flounder isolate is assigned as genotype I; genotype II includes Japanese flounder (LCDV-C) isolates; genotype III includes black rockfish (LCDV-RF) isolates; genotype IV consists of cobia and Japanese sea bass (LCDV-RC and LCDV-SB, respectively) isolates; genotype V includes painted glassfish (LCDV-CB) isolates; genotype VI for gourami; genotype VII consist of gilthead sea bream and Senegalese sole (LCDV-SA and LCDV-SSE, respectively) isolates; genotype VIII includes a largemouth bass isolate; and genotype IX is assigned for an American yellow perch isolate.
Although the first reports of LCDV date back more than 100 years, there has been little genetic data on the virus until the last few decades. Whereas the disease was previously reported from Turkey in different years (1991, 1992, 2014 and 2015), there is limited molecular characterisation of Turkish isolates (Candan 1991; Moate, Harris, and McMohan 1992; López-Bueno et al. 2016; Labella et al. 2019).
This study aimed to perform LCDV molecular detection and molecular characterisation from samples of juvenile sea bream showing lymphocystis disease symptoms collected from a sea bream farm located in the Aegean Sea.
Materials and methods
Samples and sample preparation
Thirty fish samples collected from a cage of fish displaying lymphocystis symptoms were divided into three groups with 10 fish each. Images of symptomatic fish are presented in Figure 1. Only nodules were taken from the first group and internal organs (heart, kidney, spleen, liver, digestive system) were taken from the other two groups, so three pools were created. L-15 medium with 15% FBS and antibiotic antimycotic solution (300 IU/mLpenicillin, 30 μg/mL streptomycin and 0.075 mg/mLamphotericin B.) were added at a ratio of 1:10 to pooled tissue samples and homogenised using a homogeniser (Allsheng/BIOPREP-24). Homogenates were centrifuged at 3500 rpm for 15 min at 4°C and then supernatant was collected and passed through a 0.45 µM membrane filter. Finally, filtered supernatants were stored at -80°C until nucleic acid isolation.

Nucleic acid isolation and qPCR
According to the manufacturer’s instructions, the total nucleic acid isolation was performed using Roche MagNA Pure LC and MagNA Pure LC Total Nucleic Acid Isolation Kit.
qPCR was performed using the FastStart Essential DNA Green Master kit (Roche, Germany) and a previously described primer set (Ciulli et al. 2015), LCDV qPCR F1 and LCDV qPCR R3. The reaction mixture was 20 μL total reaction volume and consisted of 10 μL Mastermix, 5 μL RNA template, 3 μL H2O and 1 μL (10 μM) of each primer.
The thermal profile used for qPCR was 95°C for 10 min, followed by 42 cycles of 95°C for 10 s, 50°C for 10 s and 72°C for 20 s. After the run, the melting curve (60-95°C) of each amplicon was examined to determine the specificity of the amplification.
Conventional PCR and sequence analysis
The PCR assay was conducted using LC1-F, LC1-R primer sets, as previously described (Kitamura et al. 2006) targeting MCP gene and the Xpert Fast Hotstart Mastermix with Dye kit (GRIPS, Portugal) according to the following program 95°C for 15 min; 45 cycles of 95°C for 15 s, 55°C for 15 s and 72°C for 15 s; and then 72°C for three min. Products were then subjected to agarose gel electrophoresis (1.5%) in TAE buffer and visualised under UV light.
Sequence analysis was performed in two directions by a commercial company (Microsynth, Switzerland). Data obtained from sequence analysis were assembled and edited using DNADynamo software.
Alignment and phylogenetic analysis
Sequences obtained in the study were downloaded from the gene bank (sequences selected from the gene bank were those whose length was equal to the sequence obtained in this study and longer ones were preferred). They were aligned by the Clustal W method using MEGA X software (Kumar et al. 2018).
Maximum Likelihood phylogenetic trees and distances between nucleotide sequences were inferred using Tamura 3- parameter model + gamma distribution (T92+G) with 1000 bootstrap replicates using MEGA X software (Tamura 1992; Kumar et al. 2018). Amino acid distances were calculated using the Jones-Taylor-Thornton model + gamma distribution (JTT+G) by MEGA-X software (Jones et al., 1992; Kumar et al. 2018).
Results
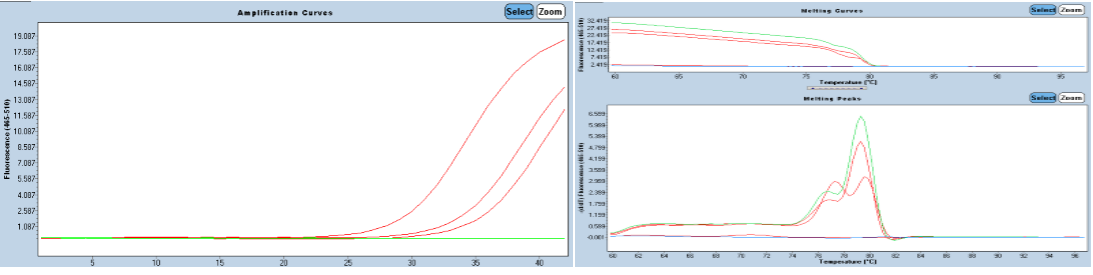
As a result of qPCR, amplification was observed in all samples and the specificity of the amplification was confirmed by melting curve analysis. Amplification, melting curve and melting peaks are presented in Figure 2. Since the studied three sample pools were collected from the same cage, sequencing was carried out with one of the three obtained sequences. The detected virus was named TR LCDV BVKE 11.09.21.

The PCR products of the partial MCP genes were detected as a 1356 base pair amplicon on the 1.5% agarose gel. As a result of editing and assembling the raw data sequence, a consensus sequence of 1263 bp was obtained. This partial sequence corresponds to 65000-66263 nucleotide genomic region of the complete genome of reference LCDV-3 virus SA9 (accession no: KX643370). This region contains between 20 and 440 amino acids of the MCP gene. The newly generated sequence has been deposited into Genbank under the accession number OM160647. All molecular studies were performed using this region.
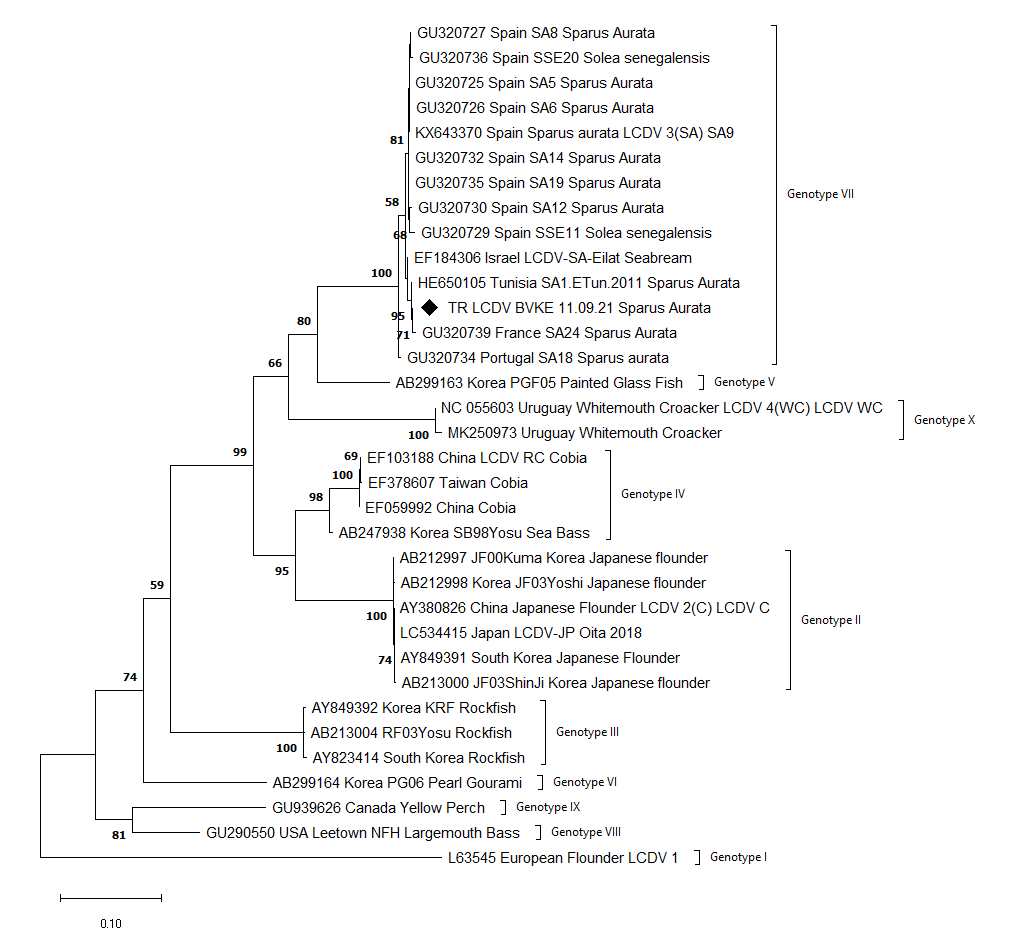
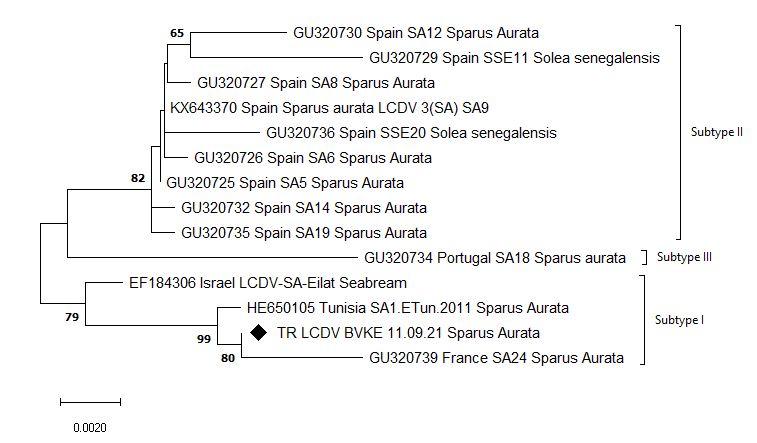
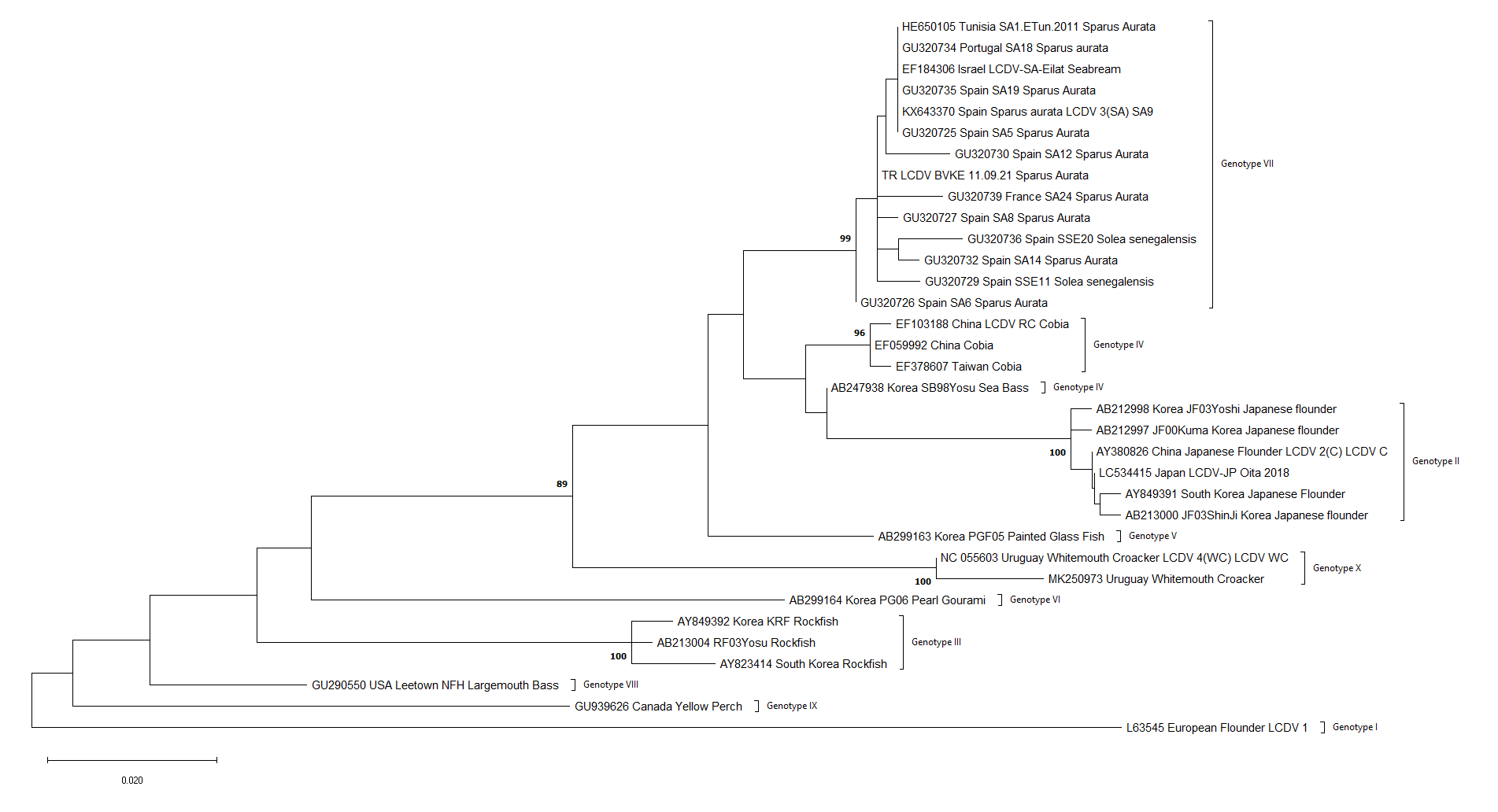
Detected virus, TR LCDV BVKE 11.09.21, was taxonomically grouped as genotype VII. Genotype VII was divided into three subtypes in the ML phylogenetic trees using nt sequences. Turkey, Israel, France and Tunisia viruses were grouped into subtype I while Spain viruses in subtype II and Portugal viruses in subtype III. The reconstructed ML phylogenetic trees are shown in Figures 3a, 3b and 3c.



The nt and aa sequence distances of TR LCDV BVKE 11.09.21 to other viruses vary between 0.16-33.43% and 0.0001-15.5%, respectively. According to the nt sequence, SA1.ETun.2011 (HE650105) reported from Tunisia was the closest, and LCDV 1 (L63545) was the most distant to TR LCDV BVKE 11.09.21. In terms of aa, SA5, SA9 and SA19 from Spain, SA1.ETun.2011 from Tunisia and SA18 from Portugal were determined as closest to TR LCDV BVKE 11.09.21, while LCDV-1 was determined as the most dissimilar. When distances between the genotypes according to the nucleotide sequences were investigated, it was determined that the closest was Genotype V with 85.8% similarity and the most distant genotype, genotype I, with 66.6% similarity. The distances between genotypes are given in Table 1.
Amino acids substitutions of between 20 and 440 of the MCP gene aa sequence were investigated using LCDV-1 (accession number: L63545) as reference. As a result of the alignment of our sequence with the reference showed that 34 aa substitutions occurred in the partial aa sequences of the MCP gene. In all sequences there were fourteen identical aa substitutions. These fourteen aa substitutions and their localisations are shown in Table 2. Among the aa sequences of viruses classified in genotype VII, 14 substitutions were observed.
Discussion
Although sea bream production in the Mediterranean basin is a relatively young industry, it has grown and developed since the 80s and 90% of the production is carried out by six countries, including Turkey (Muniesa et al. 2020). Sea bream production in Turkey ranks 3rd in total fish production, following sea bass and rainbow trout, with 110 thousand tons, according to 2020 data (TURKSTAT 2021). The Aegean Sea is the region where sea bream production is most intense in Turkey. In this study, molecular detection and characterisation of LCDV were carried out in a sea bream farm located in the Aegean Sea.
Although the mortality rate of the disease is generally low, it has been reported that in some species (Japanese flounder, red sea bream and cobia), that the mortality rate can reach 30%, and significant deaths are experienced in juvenile sea bream (Kvitt, Heinisch, and Diamant 2008; Zhan et al. 2010). In addition to high mortality rates, the inability to commercialise symptomatic fish causes economic losses (Masoero et al. 1986). The fact that the mortality rate was 20% higher than previous years in the farm where the sampling was undertaken also supports these data, and overlaps with these data with regards to LCDV causing mortality. While the signs of the disease usually occur on the skin, tail and fins, sometimes lesions can also be seen in the internal organs and it has also been reported that the virus has a wide tissue tropism in studies with asymptomatic and recovered fish (Anders 1989; Valverde et al. 2017). Although no macroscopic lesions were observed in the internal organs of fish, the virus was detected in sample pools obtained from the internal organs.
According to the phylogenetic classification studies performed targeting the major capsid protein gene, which is a relatively conserved region of iridoviruses, nine genotypes of LCDV have been described (Tidona et al. 1998; Palmer, Hogan, and van den Heuvel 2012). The LCDV-WC which was reported as the 4th species of the Lymphocystivirus by Perretta et al. (2020), exhibits a separate phylogenetic branching from the other viruses in the ML phylogenetic tree, and thus was subsequently named genotype X in this study.
In the phylogenetic tree reconstructed using nucleotide sequences, genotype VII was divided into three main branches; the first branch consisted of Israel, France, Turkey and Tunisia viruses, the second one contained only Spanish viruses and the third one which is rooted in a common node with the second, contained a Portuguese isolate. According to this branching, we suggest that this grouping reflects the geographical distribution. However, this distinction was not observed in the phylogenetic tree constructed using amino acid sequences. Moreover, when aa and nt sequences distance results were compared, a discrepancy was observed between the results. For example, the Portugal viruses (GU320734), which was the most distant to TR LCDV BVKE 11.09.21 according to the nt similarity, was amongst the most similar in terms of aa sequences. This situation reveals the necessity of investigating the molecular evolution of the virus and its circulation in the Mediterranean region, as well as investigating whether phylogenetic analyses targeting other gene regions can provide a consistent distinction between isolates.
MCP, which constitutes a large part of the virus, has been used as a target protein for creating new generation vaccines (Zheng et al. 2006; Tian et al. 2008; Zheng et al. 2010; Leiva-Rebollo et al. 2021). It was reported in these studies that DNA vaccines provide protection in related fish species. However, no study has been conducted on whether these vaccines confer cross-protection between genotypes or not. When investigating the aa sequence of the MCP, a high number of aa mutations are noted. This result suggests that cross-protection between genotypes may be minimal, but wet-lab studies are needed to confirm this. Furthermore, even if aa substitution rates among viruses grouped in genotype VII is relatively low, the vaccine’s protection created using the MCP gene of LCDV-SA against other viruses classified in genotype VII should be investigated. To our knowledge, this study is the first study in which molecular characterisation of the LCDV has been performed in Turkey. In addition, it was determined that the MCP gene of LCDV reported from sea breams from the Mediterranean basin was relatively conserved. Further studies should be conducted to investigate whether mutations in the aa sequence cause a change in the antigenic structure of the protein.